-
Naphthalenediimides with Cyclic Oligochalcogenides in Their Core
I. Shybeka, A. Aster, Y. Cheng, N. Sakai, A. Frontera, E. Vauthey and S. Matile
Chemistry - A European Journal, 26 (2020), p14059-14063


DOI:10.1002/chem.202003550 | unige:144761 | Abstract | Article HTML | Article PDF | Supporting Info
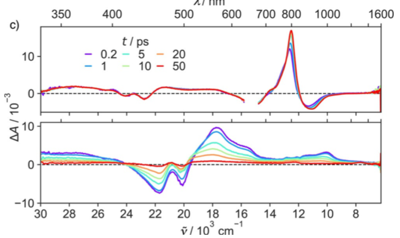
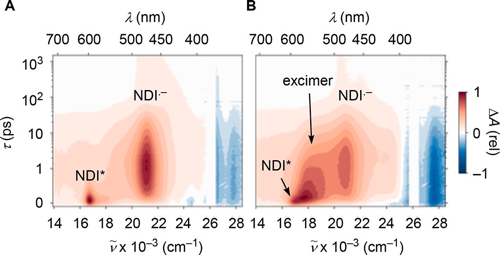
Naphthalenediimides (NDIs) are privileged scaffolds par excellence, of use in functional systems from catalysts to ion channels, photosystems, sensors, ordered matter in all forms, tubes, knots, stacks, sheets, vesicles, and colored over the full visible range. Despite this extensively explored chemical space, there is still room to discover core-substituted NDIs with fundamentally new properties: NDIs with cyclic trisulfides (i.e., trisulfanes) in their core absåorb at 668?nm, emit at 801?nm, and contract into disulfides (i.e., dithietes) upon irradiation at <475?nm. Intramolecular 1,5-chalcogen bonds account for record redshifts with trisulfides, ring-tension mediated chalcogen-bond-mediated cleavage for blueshifts to 492?nm upon ring contraction. Cyclic oligochalcogenides (COCs) in the NDI core open faster than strained dithiolanes as in asparagusic acid and are much better retained on thiol exchange affinity columns. This makes COC-NDIs attractive not only within the existing multifunctionality, particularly artificial photosystems, but also for thiol-mediated cellular uptake.